ggvolc turns the output of a differential-expression
(DE) analysis into a polished, publication-ready volcano plot. It
accepts the output of DESeq2, edgeR,
and limma directly, highlights and labels genes of
interest, combines the plot with a gt table, and can render
an interactive version with ggiraph.
The example data
The package ships with two data frames. all_genes is a
complete DESeq2-style result table; attention_genes is a
small subset of genes you might want to highlight.
data(all_genes)
data(attention_genes)
head(all_genes)
#> genes baseMean log2FoldChange lfcSE stat pvalue
#> 1 GCR1 7201.5782 2.244064 0.2004959 11.192564 4.434241e-29
#> 2 OPI10 1009.4171 -2.257454 0.2096469 -10.767889 4.880607e-27
#> 3 AGA2 249.1173 3.829474 0.3623263 10.569132 4.143136e-26
#> 4 FIM1_1376 5237.5035 2.550409 0.2560379 9.961059 2.256459e-23
#> 5 HMG1 10838.1037 2.214300 0.2229065 9.933763 2.968371e-23
#> 6 FIM1_3918 2456.8070 2.288243 0.2356228 9.711467 2.694309e-22
#> padj
#> 1 2.153711e-25
#> 2 1.185255e-23
#> 3 6.707736e-23
#> 4 2.739905e-20
#> 5 2.883475e-20
#> 6 2.181043e-19A basic volcano plot
Passing a single data frame colours every gene by significance. By
default a gene is called significant when its adjusted
p-value (FDR) is below 0.05 and its
|log2FoldChange| exceeds 1.
ggvolc(all_genes)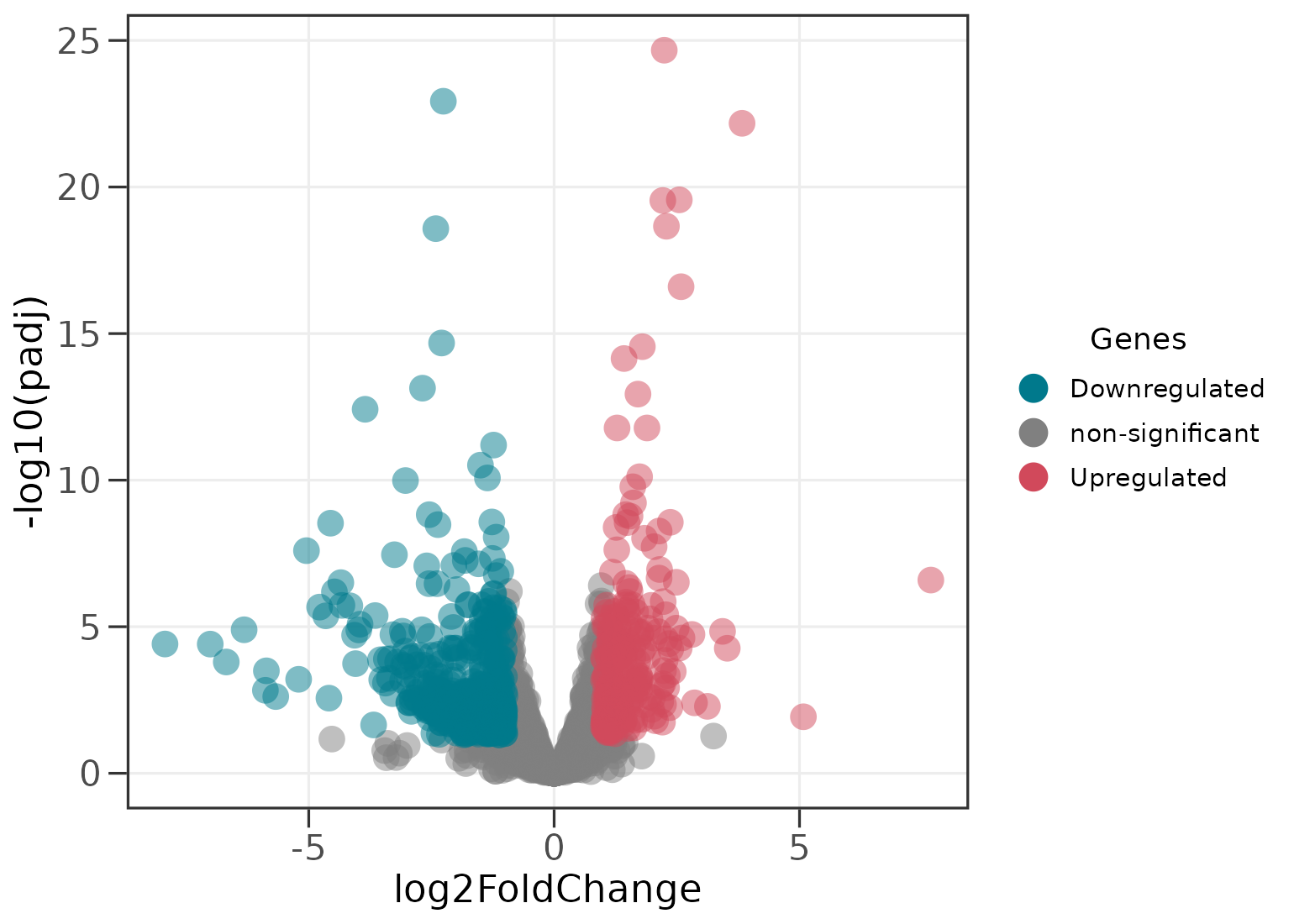
Highlighting genes of interest
Supply a second data frame and those genes are drawn with a black outline and labelled.
ggvolc(all_genes, attention_genes, add_seg = TRUE)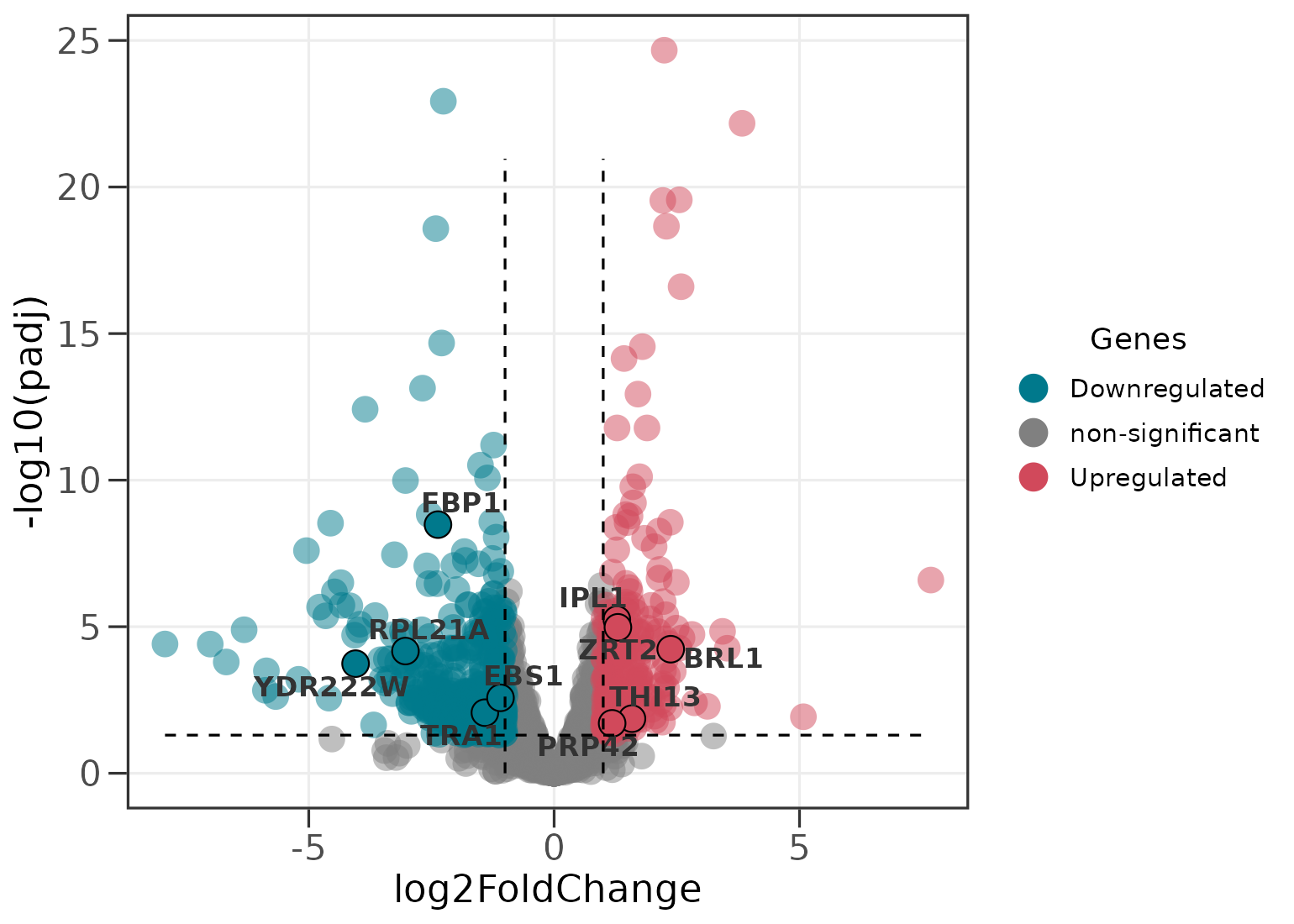
The add_seg = TRUE argument adds dashed guides at the
fold-change and significance thresholds.
Labelling the top genes automatically
You usually do not want to build a separate data frame just to label
a handful of hits. label_top picks the most significant
genes for you, and label_dir controls the direction they
are drawn from.
# the 10 most significant genes overall
ggvolc(all_genes, label_top = 10, add_seg = TRUE, title = "Top 10 hits")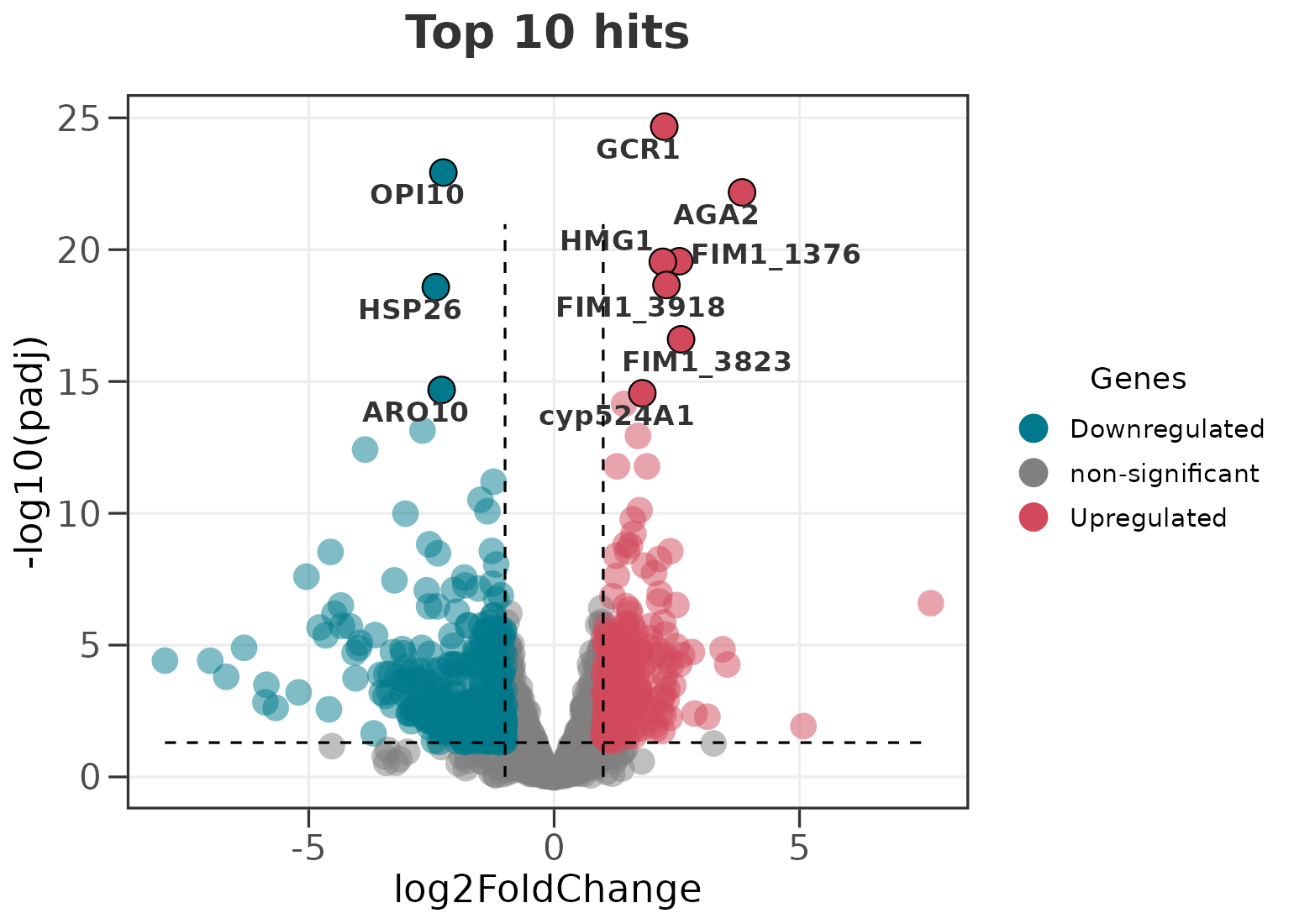
# the top 8 up- and top 8 down-regulated genes
ggvolc(all_genes, label_top = 8, label_dir = "each",
add_seg = TRUE, title = "Top 8 up + 8 down")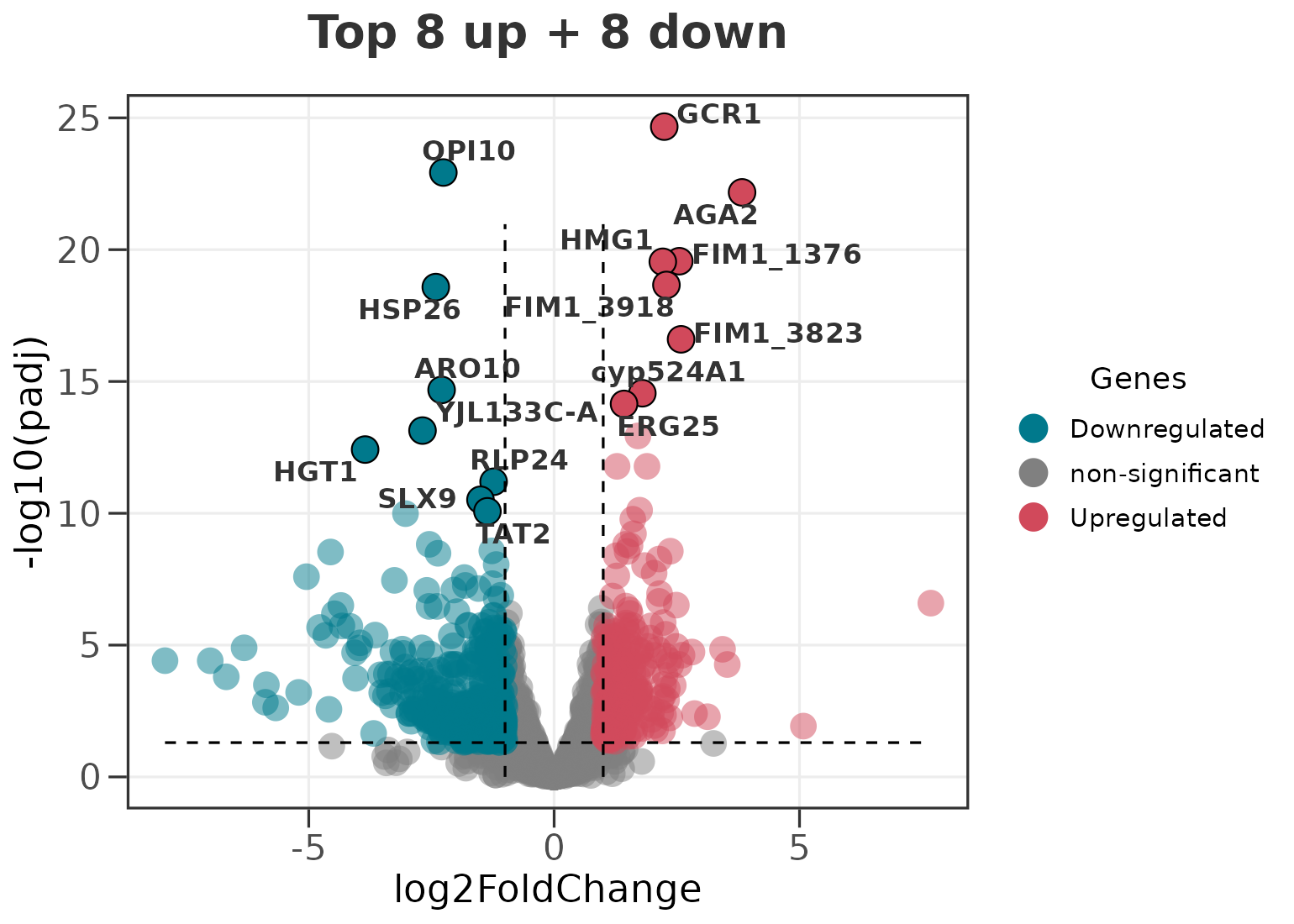
label_dir accepts "both" (the default),
"up", "down", and "each".
Calling significance on the FDR (or the raw p-value)
ggvolc() calls significance on the adjusted p-value
(padj) by default, which is the right cutoff for most DE
workflows. The y-axis and the significance line follow the choice, so
the plot stays internally consistent. Switch to the raw p-value with
sig_col = "pvalue".
ggvolc(all_genes, sig_col = "pvalue", add_seg = TRUE,
title = "Significance on the raw p-value")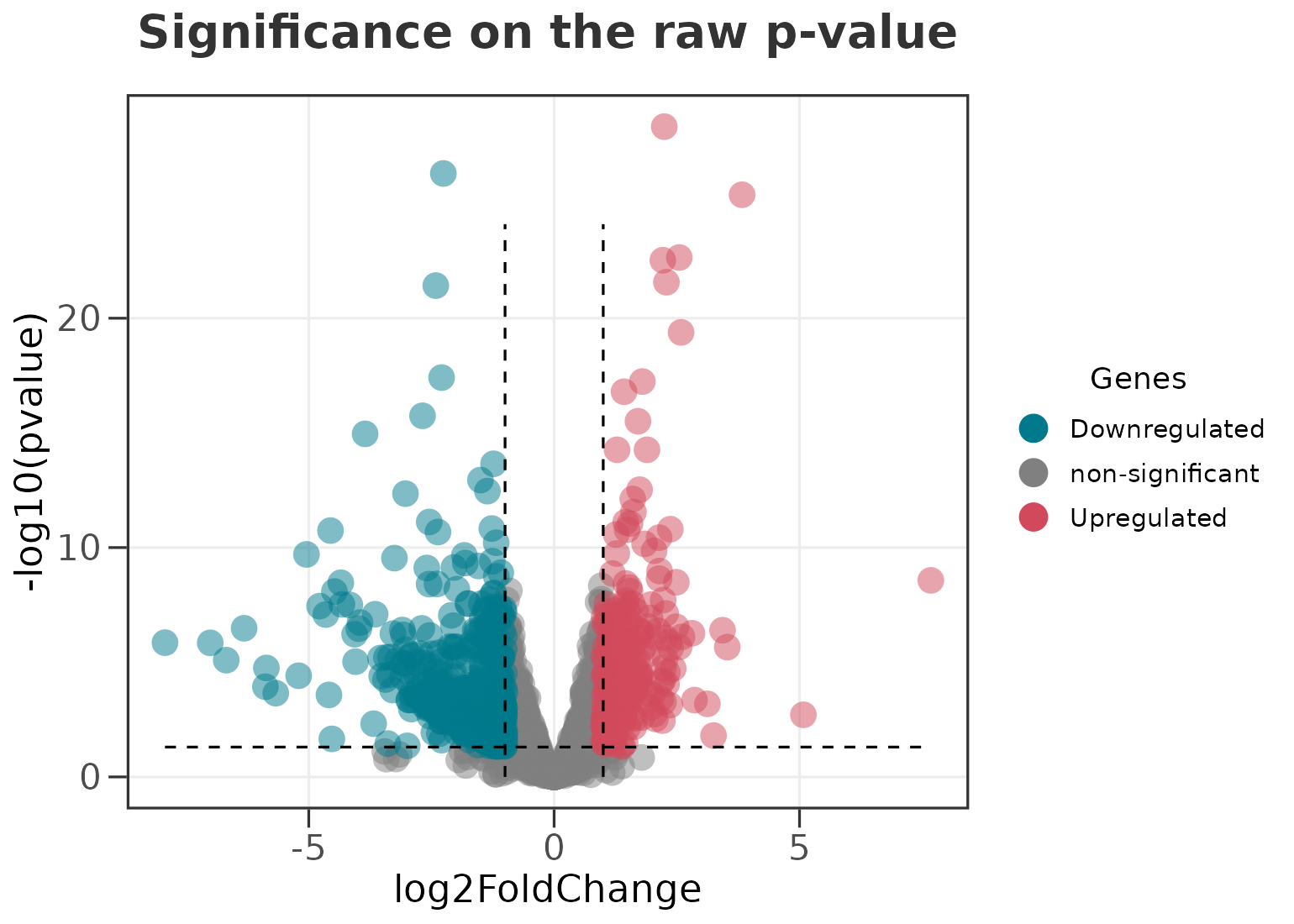
ggvolc() is also robust to p-values of exactly
0 (which DESeq2 and edgeR can emit for the strongest
genes): rather than dropping those points, their -log10
value is capped so they stay near the top of the plot.
Scaling point size
Point size can encode either the fold change or the significance.
ggvolc(all_genes, attention_genes, size_var = "log2FoldChange", add_seg = TRUE)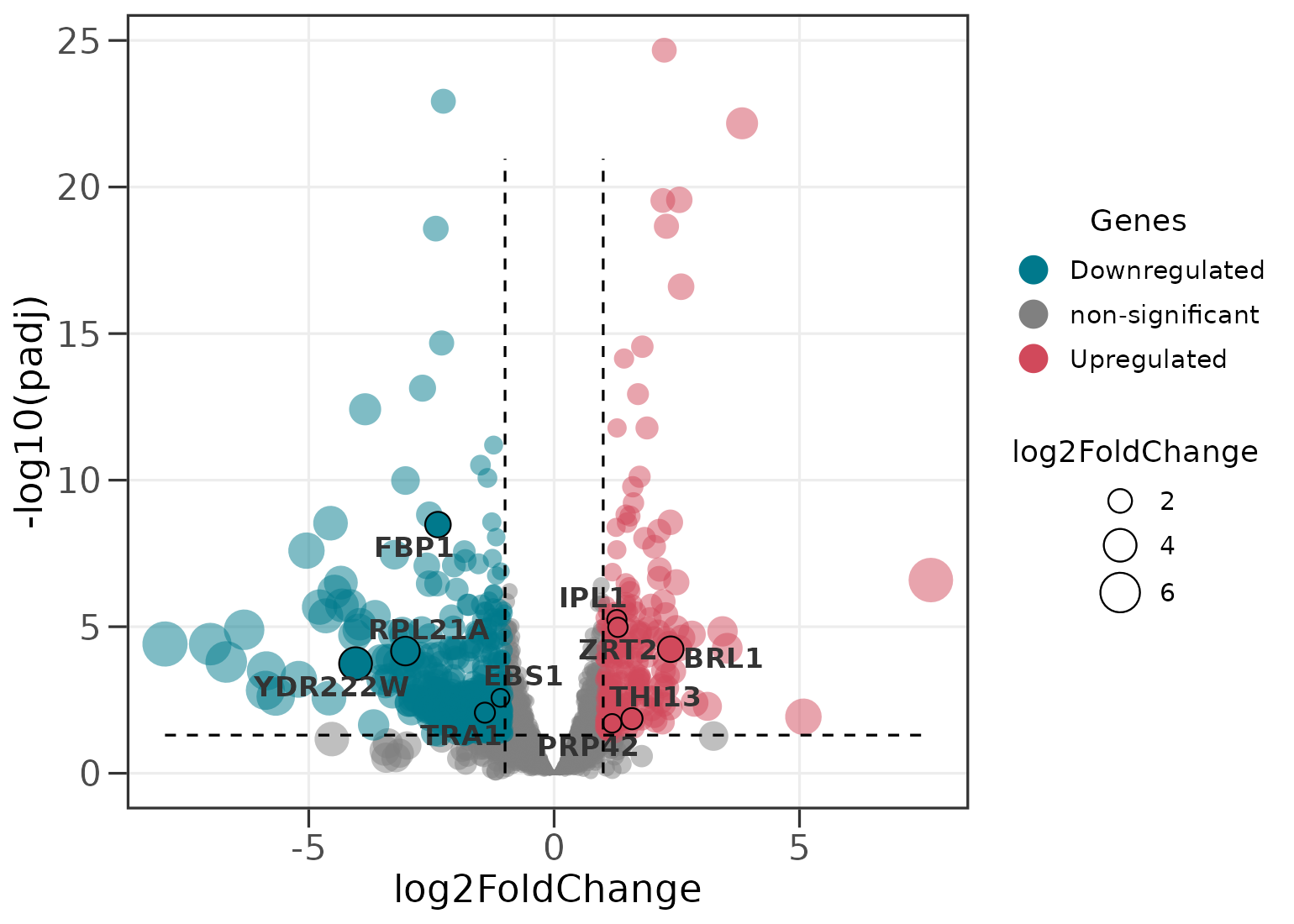
Adding a gene table
genes_table() composes the volcano plot with a
gt table of the highlighted genes using
patchwork. The result is a single object you can save with
ggplot2::ggsave().
p <- ggvolc(all_genes, attention_genes, add_seg = TRUE)
genes_table(p, attention_genes)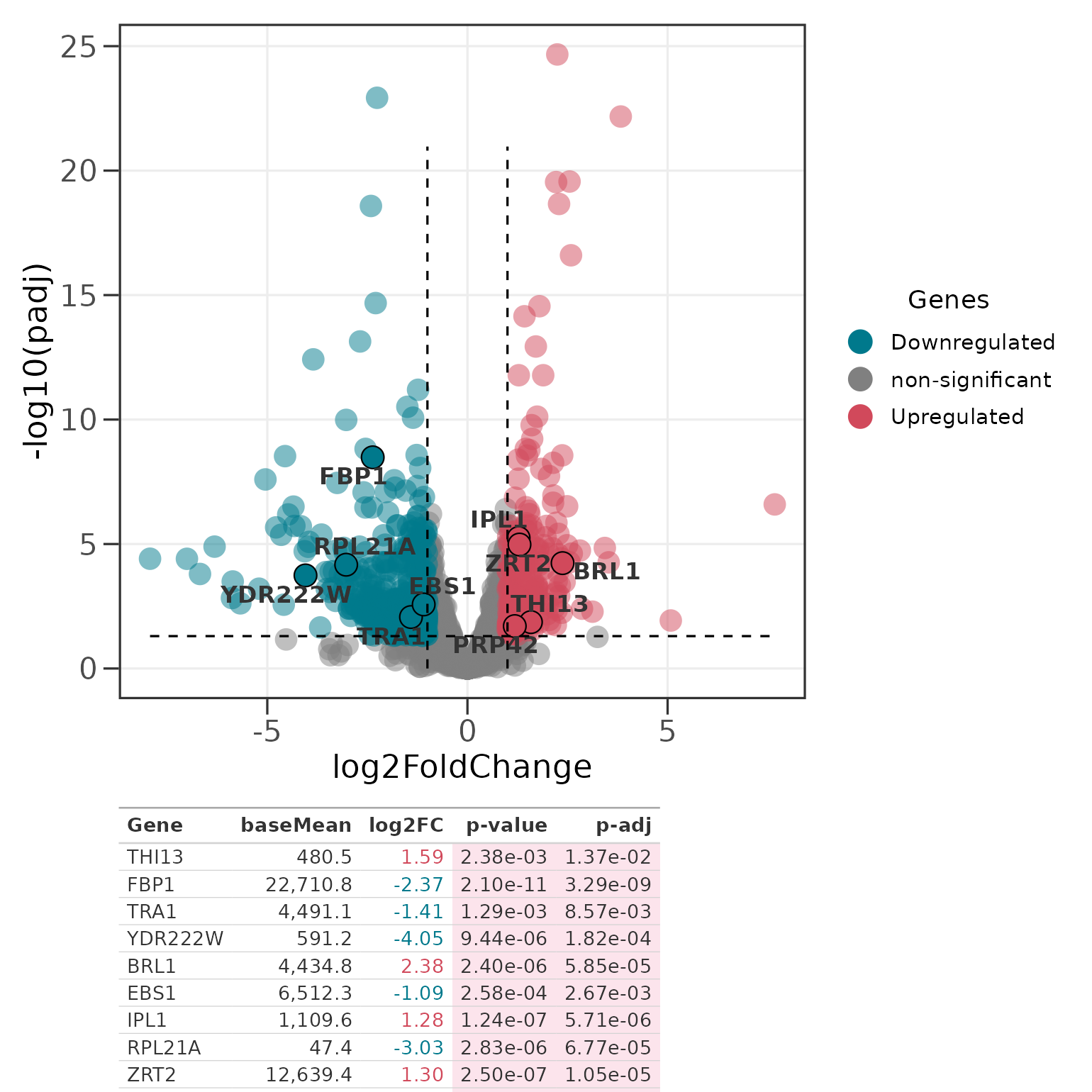
To tabulate the most significant genes without curating them
yourself, pass the full DE table and a top_n. Genes are
ranked by sig_col (padj by default, falling
back to pvalue); dir = "each" takes the top N
up- and down-regulated genes.
p_top <- ggvolc(all_genes, label_top = 10, add_seg = TRUE)
genes_table(p_top, all_genes, top_n = 10)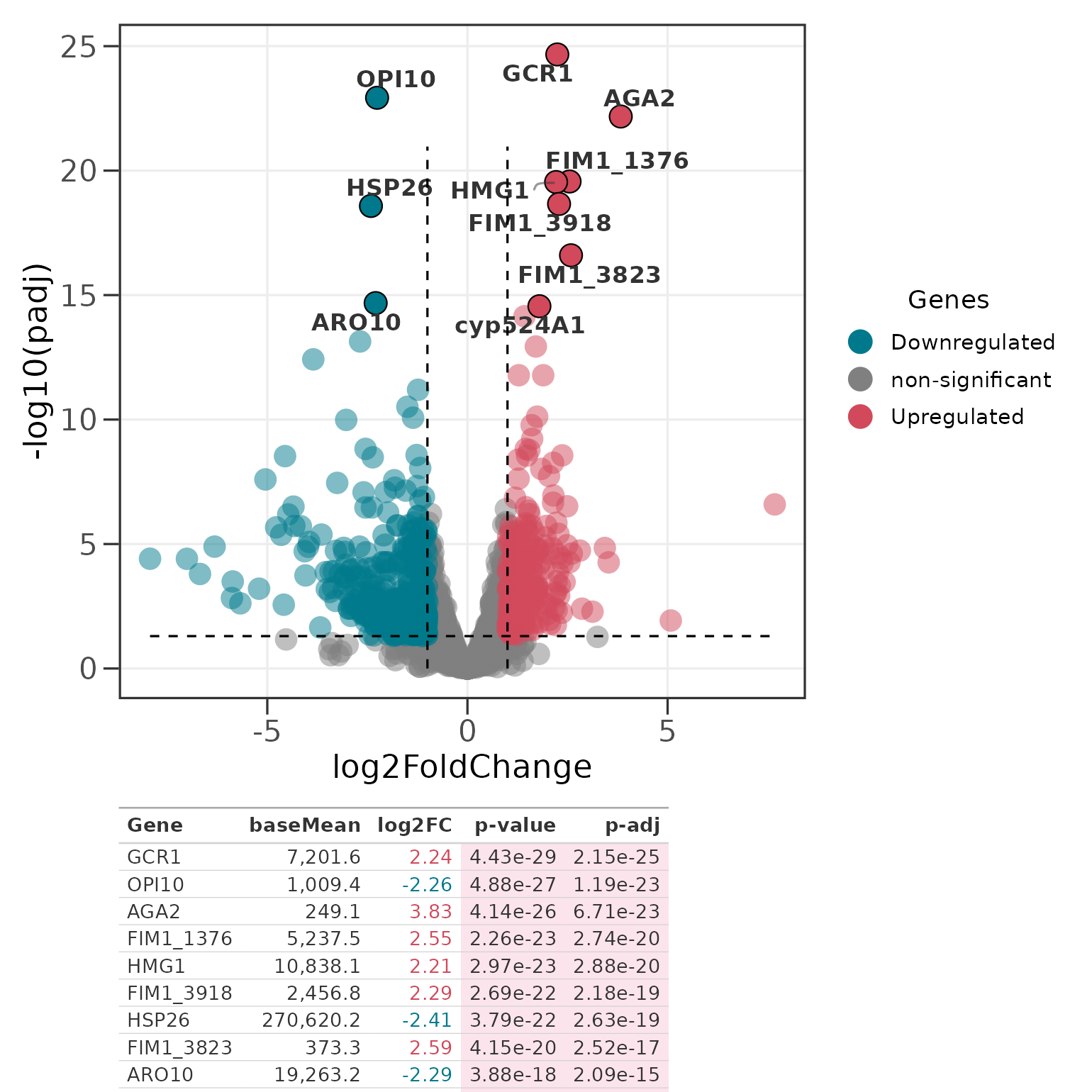
Works with DESeq2, edgeR, and limma
Column names from all three pipelines are detected and mapped
automatically, so you can pass their output straight in. Gene
identifiers held in the row names (as edgeR and limma often do) are
promoted to a genes column for you.
The package ships edger_genes, an example
topTags()-style table with the gene identifiers in the row
names, so you can try this right away:
data(edger_genes)
head(edger_genes, 3)
#> logFC logCPM PValue FDR
#> GCR1 2.244064 12.8143 4.434241e-29 2.153711e-25
#> OPI10 -2.257454 9.9807 4.880607e-27 1.185255e-23
#> AGA2 3.829474 7.9665 4.143136e-26 6.707736e-23
ggvolc(edger_genes, label_top = 8, add_seg = TRUE, title = "edgeR input")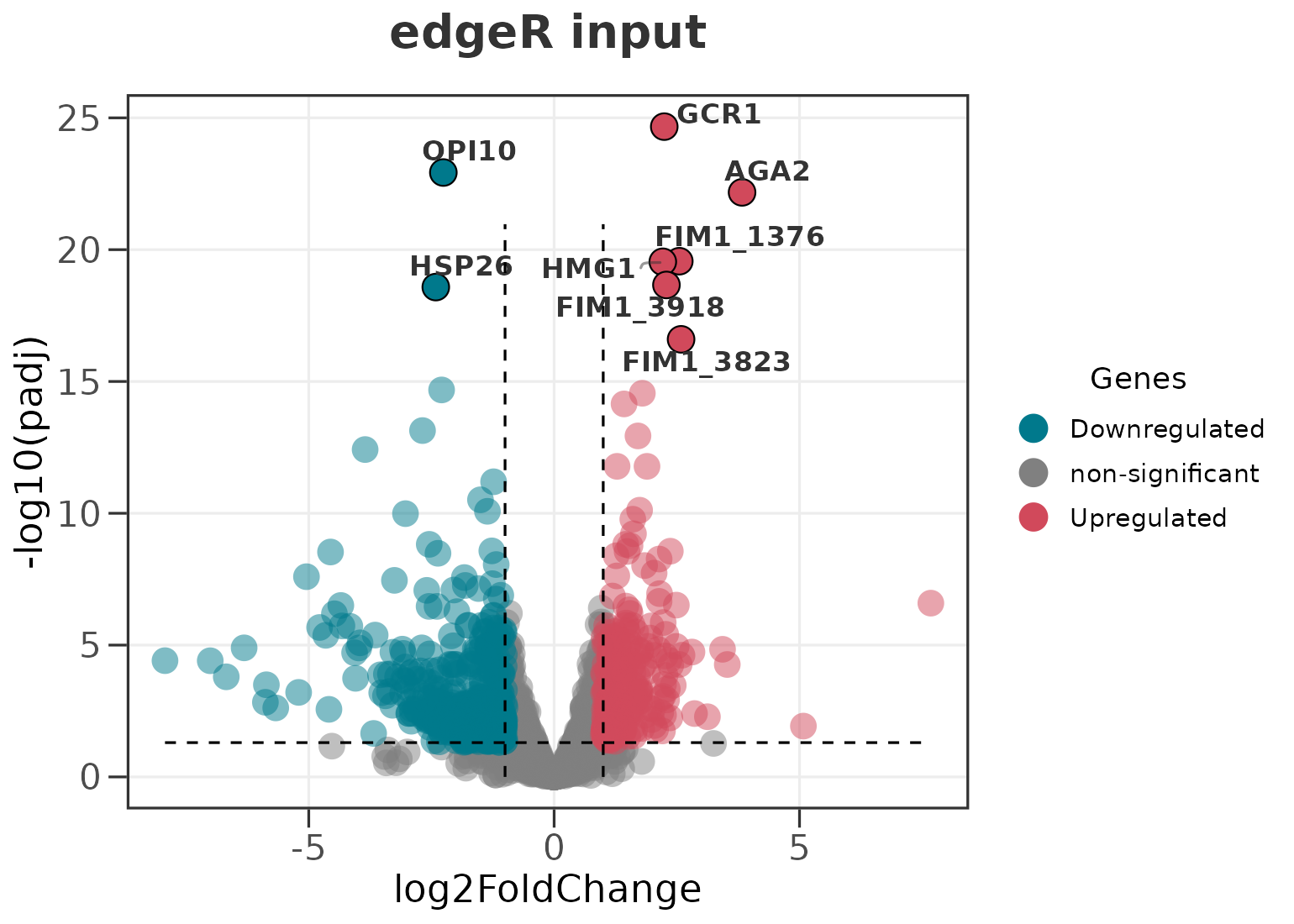
| Pipeline | Fold change | p-value | adjusted p | expression |
|---|---|---|---|---|
DESeq2 (results()) |
log2FoldChange |
pvalue |
padj |
baseMean |
edgeR (topTags()) |
logFC |
PValue |
FDR |
logCPM |
limma (topTable()) |
logFC |
P.Value |
adj.P.Val |
AveExpr |